Generalized Status Epilepticus
A seizure lasting longer than 5 mins or without return to baseline between ictal events
The longer its allowed to go on, the harder it is to treat, as the neurological tissue becomes refractory
DDx
- Structural: tumor
- Vascular: ischemia, venous sinus thrombosis
- Electrical: epileptic
- Metabolic: hypoglycemia, hypocalcemia, hyponatremia, thyrotoxicosis
- Infectious: systemic infections, CNS infections esp HSV encephalitis (prefers to affect temporal lobes, which are epileptogenic)
- Withdrawal or overdose (theophylline, demerol, some antibiotics)
- Nutritional
Tx: (Dr.A's protocol)
First, airway/breathing/circulation/accucheck (or empiric 50mL amp of D50), 100mg of thiamine, consider 1mg naloxone if you think there is a risk of opiate tox
1. Abortive - IV Ativan at 2mg increments to a max of 7-8mg; IV Valium is second line. The more benzos you give, the more you have to contend with the possibility of losing the airway and having to intubate.
2. If the seizure doesn't break immediately with the first 2mg push of IV ativan, load with fosphenytoin 15 mg PE/kg (PE= phenytoin equivalent) or 18mg/kg phenytoin and then decide whether to give more benzos.
- Note: Fosphenytoin can be pushed 3x faster than phenytoin (150mg/min vs 50mg/min). Dilantin pushed fast = risk of cardiac dysrhythmias, hemodynamic instability, tissue necrosis with extravasation. Fospheny has a lower risk of all these complications.
- Never mix dilantin with dextrose solution (increases risk of precipitation), always mix with normal saline
- If someone is allergic to dilantin, load with Vimpat ($$$) or Depakote.
3. If the seizure still didn't break after you completed loading with dilantin/fosphenytoin, then intubate and induce coma, with the goal of flatline EEG for 24-48 hours.
- Ideally, induce AND maintain with pentobarb, but its often not available on short notice
- If pentobarb unavail, induce with versed or propofol and maintain with pentobarb
- Can't maintain with versed -- it requires huge (and ever-increasing) doses to keep on for longer than 12-24 hrs, and can't maintain with propofol, due to risk of propofol infusion syndrome - high doses for prolonged time period (>1-3 days) leads to cardiac/renal failure, rhabdo, acidosis.
- Second line maintenance agent: phenobarb: very long half life ~3 days. Preferred for humans seizing 2/2 tumors. Interesting factoid: this is the agent of choice for dogs with seizures.
Other potential treatments that are less-used/more unorthodox for if everything else fails:
- ketamine
- ketogenic diet
- ECT
- plasma exchange - if the suspected etiology is autoimmune/paraneoplastic or something else plasma exchangeable
- focal surgery
Weaning off pentobarb-- goal of therapeutic levels of 2-3 AEDs of differing classes and mechanism of action (ie. dilantin/vimpat/depakote)
Long term AED: for patients that are clinically high-performing and doing well with a good EEG, can consider wean at 1-2 months, otherwise the AED cocktail should be continued
Tuesday, April 14, 2015
Monday, April 13, 2015
Ventilator management for the Neuro ICU
- Neuro patients are typically intubated for airway protection from decreasing mental status, and not for intrinsic heart or lung disease. Many neuro ICU patients have (relatively speaking) good cardiopulmonary function and reserve.
- The priorities of the neuro ICU are to optimize brain perfusion even at the cost of large insults to the other organ systems.
- Those with increased ICP are most threatened by hyponatremia and CO2 retention, and the goal will be to avoid these events - i.e. under no circumstances do we allow permissive hypercapnea in these patients, even though it may be beneficial to the lungs.
- NB about CO2 and ICPs: CO2 should not be allowed to drop any lower than 25-28, below that you start getting ischemia.
- NB#2: hypocapnea only works to lower ICP temporarily, it doesn't work forever.
Full support: A/C aka volume control
- Vent delivers preset tidal volume for machine-initiated and patient-initiated respirations
- That's problematic because it can lead to the CO2 being blown too low - i.e. in those with central hyperventilation. Remember minute ventilation = RR * TV
- If you want to enforce the vent settings, you have to really sedate people - i.e. to prevent overbreathing
- Start settings: Rate 14 (titrate to goal PCO2), TV 450 (small = lung protective), FiO2 40% (100% at intubation, titrate down to 40 asap), PEEP 5 to overcome resistance of circuit.
- Titrate PEEP and FiO2 if the sats/PO2 sucks, titrate rate and TV if pCO2 sucks.
- Other disadvantage of full support vent - diaphragm atrophy, difficulty upon weaning.
Less support: SIMV +/- pressure support
- Delivers set TV for machine-initiated respirations, delivers either no support for patient-initiated respirations (SIMV only) or delivers pressure support for patient-initiated respirations (SIMV+PS)
- At most places that I've worked at, when someone talks about "SIMV" they are talking about SIMV+PS.
- Start settings: Rate 10, TV 450, 40% FiO2, PEEP 5, PS 5.
Least support: Pressure support
- No rate, no TV, only FiO2, PEEP and PS
- All breaths are patient initiated.
- Settings: 40% FiO2, PEEP 5, PS 5
- Neuro patients are typically intubated for airway protection from decreasing mental status, and not for intrinsic heart or lung disease. Many neuro ICU patients have (relatively speaking) good cardiopulmonary function and reserve.
- The priorities of the neuro ICU are to optimize brain perfusion even at the cost of large insults to the other organ systems.
- Those with increased ICP are most threatened by hyponatremia and CO2 retention, and the goal will be to avoid these events - i.e. under no circumstances do we allow permissive hypercapnea in these patients, even though it may be beneficial to the lungs.
- NB about CO2 and ICPs: CO2 should not be allowed to drop any lower than 25-28, below that you start getting ischemia.
- NB#2: hypocapnea only works to lower ICP temporarily, it doesn't work forever.
Full support: A/C aka volume control
- Vent delivers preset tidal volume for machine-initiated and patient-initiated respirations
- That's problematic because it can lead to the CO2 being blown too low - i.e. in those with central hyperventilation. Remember minute ventilation = RR * TV
- If you want to enforce the vent settings, you have to really sedate people - i.e. to prevent overbreathing
- Start settings: Rate 14 (titrate to goal PCO2), TV 450 (small = lung protective), FiO2 40% (100% at intubation, titrate down to 40 asap), PEEP 5 to overcome resistance of circuit.
- Titrate PEEP and FiO2 if the sats/PO2 sucks, titrate rate and TV if pCO2 sucks.
- Other disadvantage of full support vent - diaphragm atrophy, difficulty upon weaning.
Less support: SIMV +/- pressure support
- Delivers set TV for machine-initiated respirations, delivers either no support for patient-initiated respirations (SIMV only) or delivers pressure support for patient-initiated respirations (SIMV+PS)
- At most places that I've worked at, when someone talks about "SIMV" they are talking about SIMV+PS.
- Start settings: Rate 10, TV 450, 40% FiO2, PEEP 5, PS 5.
Least support: Pressure support
- No rate, no TV, only FiO2, PEEP and PS
- All breaths are patient initiated.
- Settings: 40% FiO2, PEEP 5, PS 5
Wednesday, April 1, 2015
Eosinophilic Granuloma (Langerhans Cell Histiocytosis)
Introduction and Nosology
Langerhans Cell Histiocytosis (LCH) is a rare, heterogenous illness characterized by the proliferation of dendritic cells with Langerhans cell morphology. “LCH” refers to a spectrum of disease, from a localized lesion to diffuse multiorgan pathology. [1-3] LCH can affect any organ system; the most commonly affected are the skeletal system (80%), skin (33%), and pituitary (25%); others include the liver, spleen, lungs, and brain.[4] The most common named subtype, Eosinophilc Granuloma (EG), refers to a benign, localized LCH, most commonly of bone [2] Other named subtypes include Hand-Schuller-Christian disease, a multifocal LCH classically characterized by exophthalmos, diabetes insipidus, and osteolytic skull lesions and Letterer-Siwe disease, a diffuse systemic LCH clinically manifested by skin rash, hepatosplenomegaly, and pancytopenia. [5, 6] In 1953, Lichtenstein grouped all 3 of these diseases under the name “histiocytosis X”, a term which has since been replaced by Langerhans Cell Histiocytosis. [5, 7]
Epidemiology and Clinical Presentation
Eosinophlic Granuloma (EG) that affects the bone is the most common subtype of LCH, representing an estimated 60-80% of cases; it can be single or multifocal and most commonly affects the calvarium but can also present in the vertebrae, ribs, long bones, and mandible [6, 8, 9] EG primarily affects children under the age of 15 with an estimated incidence of less than 1 per 100,000.[1, 3]
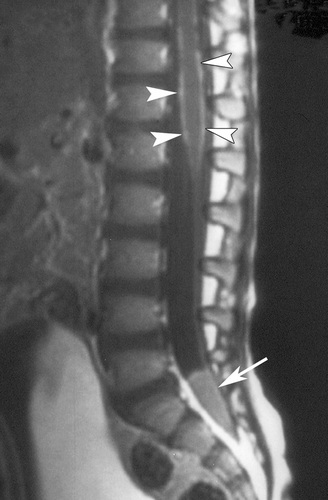
Clinically, EG presents as a painful soft tissue mass; radiography demonstrates sharply demarcated osteolytic lesions of the underlying bone. [2, 8] EG of the skull presents as a gradually enlarging scalp mass.[2] EG of the vertebral body in children most commonly presents with pain; neurologic deficit is uncommon even in cases of progression to vertebra plana. [10] [11] EG of the orbital bone may present as proptosis with an accompanying mass mimicking malignancy. [12] EG may even occur in the brain parenchyma with a tumor-like appearance; these lesions most commonly appear in the hypothalamic-pituitary axis and present with endocrinopathies. [13] Radiographically, LCH lesions are avidly contrast enhancing on both CT and MR, and demonstrate increased uptake in FDG-PET. [14] They appear hypointense on T1 MRI and iso to hyperintense on T2. [3, 15]
Etiology and Pathophysiology
LCH is characterized by the proliferation of a myeloid-derived precursor dendritic cell with the characteristics of Langerhans cells (LC). [16] [17] One theory of the pathophysiology of LCH is based on recently published data that LCH cells express CD40 at high levels, leading to the overactivation of CD40L+ T-lymphocytes. The subsequent release of cytokines leads to further recruitment of LC progenitor cells, as well as to local destruction of bone, fibrosis, and necrosis. [18-20]
The central debate of the pathogenesis of LCH is whether it is a reactive immune response or a neoplasm. Support for the neoplastic theory include the monoclonality of the pathologic cells (although this could also be seen with reactive immune processes) [21] [20], the up regulation of pro-cell proliferation and anti-apoptotic proteins (c-myc, h-ras, Bcl-2) in LCH samples, [22] the up regulation of Ki-67 (a marker of proliferation), [22] [23] the finding of activating BRAF mutations in a majority of samples and the clinical response of BRAF V600E+ disease to vemurafenib. [24] [25] Support for the theory that LCH represents a dysfunctional immune response include data showing the up-regulation of genes that lead to T-cell activation and recruitment in LCH lesions, [26] an increase of T-regulatory cells in the blood of LCH patients in comparison with controls, [27], and a lack of evidence of gross genomic or karyotypic aberration in LCH biopsy samples. [28] Of note, no antigenic trigger has ever been identified and recent evidence has mounted against a viral etiology. [29, 30]
Workup & Diagnosis
The differential for an osteolytic soft tissue mass is broad, and includes neoplastic processes: metastatic lesions, primary bone tumors such as osteosarcoma or Ewing’s sarcoma, neuroblastoma, rhabdomyosarcoma, lymphoma, PNET. Infections such as osteomyelitis or abscess. Fibrous dysplasia, cystic lesions (aneurysmal bone cyst, dermoid cyst), vascular processes (venous lakes, hemangioma, angiomatosis), and developmental anomalies (encephalocele).
Tissue biopsy is necessary for definitive diagnosis: Histopathologically, LCH lesions show a proliferation of LC-type cells in a milieu of lymphocytes, macrophages, and eosinophils. In 1987, the histiocyte society established diagnostic criteria for LCH: the identification of CD1a on immunohistochemistry or of Birbeck granules on electron microscopy. [5] Immunhistochemistry for Langerin, a protein enriched in and necessary for the formation of Birbeck granules, is an acceptable modern alternative to electron microscopy. [4, 20] Once mysterious, recent work suggests Birbeck granules are part of the eadosomal recycling system, and may be involved with loading CD1a (a protein similar to MHC-I) with glycoprotein antigens for presentation to T-cells. [31]
Workup should include a systemic survey to identify any other potential sites of involvement, as management recommendations and prognosis vary depending on the number and type of organs involved in LCH.[32] The Euro Histio network published guidelines for the workup of LCH in 2013 based on the available review of the literature, and recommend the following labs: CBC with differential, CMP (electrolytes, BUN/creatinine and liver function tests), ESR, coagulation studies (PT/INR, PTT, fibrinogen), chest x-ray and radiographic skeletal survey. [4] The histiocyte society recommends the same labs, with the addition of urine specific gravity and abdominal ultrasound.[33] Endocrine labs (FSH/LH, TSH, GH, cortisol) are indicated if there is any suspicion of pituitary involvement. MRI is useful for the detection of bone marrow or soft tissue involvement, and a fast whole-body T2 STIR protocol is one possible screening test. [34, 35] PET has been shown to be superior to bone scans for lesion detection, but exposes children to much higher radiation doses [35]. A recent study by Mueller et al found MRI had a higher sensitivity (81%) but FDG-PET had a higher specificity (76%) for the detection of LCH lesions. [36]
Treatment
Solitary EG of the calvarial vault without invasion into neurological structures has a good prognosis. Historically managed with surgical curettage or excision, [4] [17] [37] recent reports by Oliveira et al (n=4) and by De Angulo et al (n=8) found that these lesions fully resolved without intervention after 6-19 months. [17] [38] The major limitation of these studies is the lack of diagnostic certainty in the absence of tissue analysis. There is no class I evidence for the management of these lesions, and treatment decisions must be made on a case by case basis. On the one hand, overly aggressive surgical excision may be associated with prolonged healing and deformity, particularly with large lesions; the European Histiocyte Society recommends avoidance of surgical excision in lesions > 5 cm in children. [4] On the other hand, observation alone may be a risky decision, as the differential diagnosis of LCH includes aggressive cancers and infection, and the disease may progress to pathologic fracture or encroachment on neurological structures. [39] One middle ground option is to perform a biopsy or partial resection for definitive diagnosis, followed by observation or a conservative treatment option- such as intralesional injection of steroids or inteferon, or systemic treatments with indomethacin or bisphosphonates. These treatments have been shown to be efficacious in the literature, [40] [41] [42] however the results must be interpreted in the context of a disease that is potentially self-resolving.
The histiocyte society recommends more aggressive treatment in cases of multifocal bone disease or disease that involves “CNS-risk” sites (odontoid peg, vertebrae with intraspinal soft tissue extension, facial bones, skull base, orbit, oral cavity). [20] [33] In these types of LCH, the risk of recurrence is high (30-50%) as is the risk of disease invasion into neurological tissue or the development of neurological sequeale (40%) such as endocriniopathies, diabetes insipidus, and parenchymal brain disease. [33] For LCH of this type, the histiocyte society recommends systemic treatment with prednisone and vinblastine for 12 months. [33] There is no class I evidence or guideline recommendations regarding surgical treatment for these lesions. Historically, lesions that invade the dura or compress neurological tissues and that are located in accessible sites have undergone surgical excision. [6, 43] [44] In cases where surgical excision would be impossible or excessively morbid, systemic chemotherapy and/or injections of steroids or interferon may be a preferential alternative. [39] In the past, radiation therapy was used as an adjunctive treatment, however it is no longer recommended except in emergent cases of compression of critical neurological structures, such as the optic nerve or spinal cord, out of concern for the possible effects of radiotherapy on the developing brain and spine. [4] [20]
LCH of the spine is rare. Treatment patterns in the literature have varied from observation to complete surgical excision with fixation to radiotherapy. Bertram et al in a review of the literature of spine EG (n=53) found that most cases resolved without treatment, and recommend immobilization and observation only in cases without spinal instability or neurological deficit. [10] Similar findings have been reported by Raab et al (n=14) and Yeom et al (n=23). [45, 46] LCH spares the endochondral plates, and so recovery of vertebral heights is possible if the endochondral tissues are not disturbed with surgery or radiation. [45] [37] Surgical excision, followed by segmental fusion and internal fixation, is indicated in cases of spinal instability, neurological deficit from compression of the spinal cord or spinal nerves, or in patients unable or unwilling to comply with prolonged immobilization or external bracing. [37] As in cranial lesions, radiotherapy is no longer recommended for spine LCH except in emergent cases of spinal cord compression. [4] [20]
Prognosis
The prognosis for LCH is good. In a large study from South Korea (n=603), 5-year overall survival in those with single-system LCH was 99.8%, for multisystem LCH without risk-organ involvement was 98.4%, for multisystem LCH with risk-organ involvement was 77%. [47] Similar findings have been found by other groups in Japan (n=91) and Italy (n=121). [48] [49] In a large study from the Mayo Clinic (n=263), recurrence rates in those with single-bone LCH was 7%, and the appearance of new bone lesions was 14%. [44] As aforementioned, the Histiocyte Society reports a high rate of recurrence (30-50%) among those with “CNS-risk” lesions. [33]
Wednesday, March 11, 2015
Neurofibroma
General:
- neurofibromas are also a neoplastic proliferation of schwann cells, but the tumors are more heterogenous -- contain fibroblasts, perineural cells, residual axons
- while schwannomas tend to be well circumscribed tumors that push nerves to the side, neurofibromas are non-encapsulated and more invasive
- For more details see blog post from oct 8, 2014
- Associated with NF1 - in these patients, can transform to MPNST
Variants:
- localized soft tissue tumor
- diffuse neurofibroma can grow into large lesion
- plexiform variant - expands nerve fascicles
Path:
(from Dr.Pytel's CNS tumors lecture)

On imaging, can be virtually indistinguishable from schwannoma
MPNST
General:
- may look like high grade sarcoma
- 50% arise from plexiform neurofibroma in pt with NF1
- 5% of NF patients develop a MPNST
Path:

Many nuclei, cellular spindle cell tumor, lots of mitotic activity (contrast with path image above)
source: Dr.Pytel's lecture
General:
- neurofibromas are also a neoplastic proliferation of schwann cells, but the tumors are more heterogenous -- contain fibroblasts, perineural cells, residual axons
- while schwannomas tend to be well circumscribed tumors that push nerves to the side, neurofibromas are non-encapsulated and more invasive
- For more details see blog post from oct 8, 2014
- Associated with NF1 - in these patients, can transform to MPNST
Variants:
- localized soft tissue tumor
- diffuse neurofibroma can grow into large lesion
- plexiform variant - expands nerve fascicles
Path:
(from Dr.Pytel's CNS tumors lecture)
- abundant wavy collagen deposits, "carrot shavings"
On imaging, can be virtually indistinguishable from schwannoma
MPNST
General:
- may look like high grade sarcoma
- 50% arise from plexiform neurofibroma in pt with NF1
- 5% of NF patients develop a MPNST
Path:
Many nuclei, cellular spindle cell tumor, lots of mitotic activity (contrast with path image above)
source: Dr.Pytel's lecture
Tuesday, March 10, 2015
Schwannoma
General
- Peripheral nerve sheath tumors
- 8th nerve typically vestibular branch (rarely other CN), nerve roots, peripheral nerves
- Other nerve sheath tumors: neurofibromas, MPNST
- Bilateral = NF2
- Rarely undergo malignant transformation
Path
- Gross: well-circumscribed, extra-axial
- Source: Dr.Pytel's CNS tumors lecture

Antoni A and antoni B

Verucay bodies - cells without nuclei
Imaging:
 acoustic
acoustic

 source: headneckbrainspine
source: headneckbrainspine
General
- Peripheral nerve sheath tumors
- 8th nerve typically vestibular branch (rarely other CN), nerve roots, peripheral nerves
- Other nerve sheath tumors: neurofibromas, MPNST
- Bilateral = NF2
- Rarely undergo malignant transformation
Path
- Gross: well-circumscribed, extra-axial
- Source: Dr.Pytel's CNS tumors lecture
Antoni A and antoni B
Verucay bodies - cells without nuclei
Imaging:
source: radiopaedia
Monday, March 9, 2015
Medulloblastoma
General
- from cells resembling immature neurons of cerebellum
- Most common malignant/high grade tumor of children - also most common posterior fossa tumor of children (30-40%)
- Mean age of appearance - 9 years. When they occur in adults (rare) typically 3rd-4th decade, better prognosis
- May recur locally or spread in CSF - drop mets in spinal cord, or explants in hemispheres.
Prognosis
- Heterogenous group of tumors - prognosis varies widely depending on molecular signaling.
- Tx: surgical resection, chemo, whole brain and spine RT to manage potential mets/prevent spread.
- 5-year survival 50-70%, but with high morbidity 2/2 RT - i.e. short stature due to hypothalamic and spinal bony RT, secondary malignancy.
Path
- small blue cell tumor - sheets of undifferentiated cells. Sometimes rosettes.

 - sheets of blue cells
- sheets of blue cells
Neuronal lineage - stain + with neuronal markers like synaptophysin and NeuN (nuclear marker of neurons)
Imaging:
(source: radiopaedia, this paper)
- CT bright - may have significant cystic/calcific components
- T1 dark
- 90% enhance, usually heterogenously


General
- from cells resembling immature neurons of cerebellum
- Most common malignant/high grade tumor of children - also most common posterior fossa tumor of children (30-40%)
- Mean age of appearance - 9 years. When they occur in adults (rare) typically 3rd-4th decade, better prognosis
- May recur locally or spread in CSF - drop mets in spinal cord, or explants in hemispheres.
Prognosis
- Heterogenous group of tumors - prognosis varies widely depending on molecular signaling.
- Tx: surgical resection, chemo, whole brain and spine RT to manage potential mets/prevent spread.
- 5-year survival 50-70%, but with high morbidity 2/2 RT - i.e. short stature due to hypothalamic and spinal bony RT, secondary malignancy.
Path
- small blue cell tumor - sheets of undifferentiated cells. Sometimes rosettes.
- "rosettes"
Neuronal lineage - stain + with neuronal markers like synaptophysin and NeuN (nuclear marker of neurons)
Imaging:
(source: radiopaedia, this paper)
- CT bright - may have significant cystic/calcific components
- T1 dark
- 90% enhance, usually heterogenously
- medullo with leptomeningeal spread along anterior pons
- classic medullo - emerging from cb vermis
- leptomeningeal mets.
Sunday, March 8, 2015
CNS Lymphoma
General
- Primary Sporadic extranodal lymphoma (older patients) - usually EBV-neg - no extra-CNS disease
- Primary CNS lymphoma of immunocompromised patients - can occur at any age - usually EBV-associated
- Secondary CNS lymphoma = mets from primary lymphoma elsewhere, most common - 2/3 leptomeningeal/subependymal disease
- Single/multiple lesions, or diffuse infiltrate
- Mostly large B-cell lymphomas
- Tx with intrathecal chemo and RT
- Poor prognosis
Path:
 (source: Dr.Pytel's CNS tumors lecture)
(source: Dr.Pytel's CNS tumors lecture)
- no processes emanating from cells
Imaging:
- Data and images sourced from this AJNR paper
- Immunocompetent: usually homogenously enhancing (90%), ring-enhancing (0-13%)
- AIDS patients - heterogenously enhancing 75% of the time


General
- Primary Sporadic extranodal lymphoma (older patients) - usually EBV-neg - no extra-CNS disease
- Primary CNS lymphoma of immunocompromised patients - can occur at any age - usually EBV-associated
- Secondary CNS lymphoma = mets from primary lymphoma elsewhere, most common - 2/3 leptomeningeal/subependymal disease
- Single/multiple lesions, or diffuse infiltrate
- Mostly large B-cell lymphomas
- Tx with intrathecal chemo and RT
- Poor prognosis
Path:
- no processes emanating from cells
Imaging:
- Data and images sourced from this AJNR paper
- Immunocompetent: usually homogenously enhancing (90%), ring-enhancing (0-13%)
- AIDS patients - heterogenously enhancing 75% of the time
- homogenously enhancing, can be dural-based
- AIDS related disease - can be heterogenous, ring-enhancing
Subscribe to:
Posts (Atom)